I have a vector of p-values that need correcting for multiple comparisons; based on reading Noble (2009), I am attempting to apply a Benjamini-Hochberg FDR-based correction.
My vector (sorted in ascending order for ease of visualisation) is:
ps <- c(0.019, 0.022, 0.023, 0.023, 0.025, 0.025, 0.027, 0.028, 0.029, 0.030, 0.030, 0.030, 0.031, 0.033, 0.034, 0.035, 0.036, 0.037, 0.037, 0.039, 0.051, 0.060, 0.063, 0.065, 0.085, 0.110, 0.170, 0.196, 0.241, 0.316, 0.318, 0.325, 0.694)
When I run the R commmand
p.adjust(ps, method="BH")
I get the output:
0.06426316 0.06426316 0.06426316 0.06426316 0.06426316 0.06426316 0.06426316 0.06426316 0.06426316 0.06426316 0.06426316 0.06426316 0.06426316 0.06426316 0.06426316 0.06426316 0.06426316 0.06426316 0.06426316 0.06435000 0.08014286 0.08937500 0.08937500 0.08937500 0.11220000 0.13961538 0.20777778 0.23100000 0.27424138 0.33515625 0.33515625 0.33515625 0.69400000
This output clearly has certain values that are over-represented. Given that they are the same to 8 decimal places, I don't think it is likely to happen by chance. If I plot the raw and adjusted values, they both follow the same topography but the adjusted values plateau whenever the raw values have a small slope.
plot(ps~order(ps))
points(p.adjust(ps, method="BH")~order(ps), add=TRUE, col="red")
Generally, it seems that when a raw p-value has a difference <0.01 from the next-highest value, the corrected values come out identical. I can't understand why, though; I've read around and dug into the p.adjust code, but can't think what would cause this effect. Any insight would be greatly appreciated!
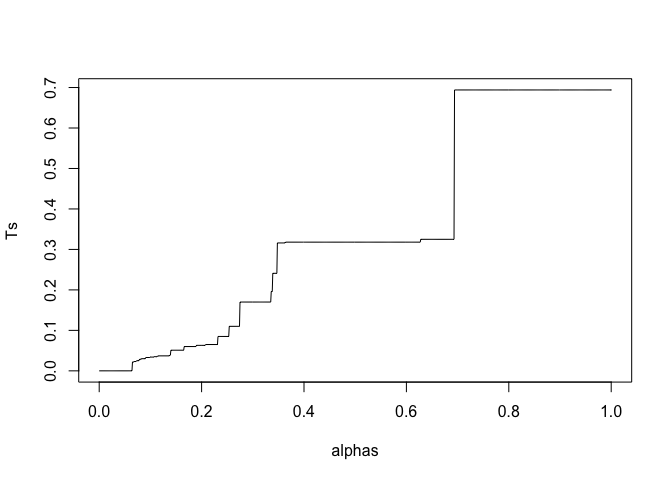
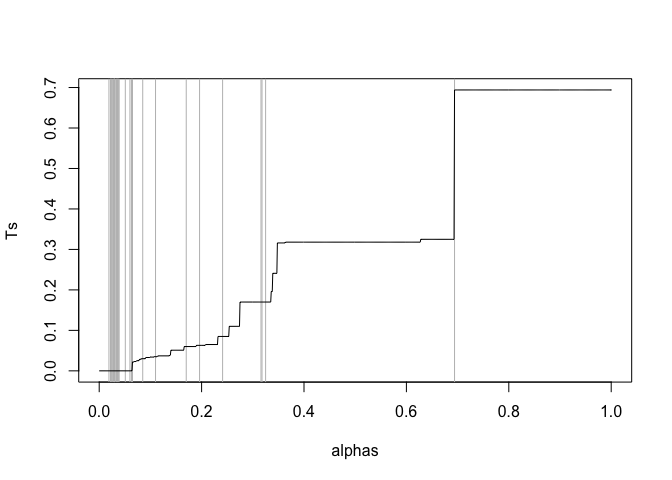 These will get rejected at the same threshold and correspond to the same smallest $\alpha$. This $\alpha$ is your q-value, so it's OK and pretty much inevitable to get repeated q-values.
These will get rejected at the same threshold and correspond to the same smallest $\alpha$. This $\alpha$ is your q-value, so it's OK and pretty much inevitable to get repeated q-values.