I am currently working on adjusting p-values in R using the False Discovery Rate (FDR) method and have encountered some confusion regarding the basic concepts of adjusted p-values and q-values. Adjusted p-values result from applying a correction method to control for multiple testing.
Indeed in https://genome.ucsc.edu/goldenPath/help/qValue.html, it's stated that: 'The q-value is an analog of the p-value that incorporates multiple testing correction. The q-value is defined as the minimum false discovery rate at which an observed score is deemed significant.'
Otherwise, read through a relevant answer https://support.bioconductor.org/p/49864/ , which emphasizes that adjusted p-values and q-values are similar but not quite the same.
I understand the algorithm for calculating q-values, and I understand that repeated values might be encountered. However, I am still seeking clarification on the differences between adjusted p-values and q-values, if any.
To illustrate my point, I'm using a simple example in R. Imagine that I have the p-values of a two sample t-test with alpha=0.05 conducted on the same individuals for 43 variables:
set.seed(6655)
p_values <- abs(rnorm(43,mean = 0, sd=0.25))
adjusted_p_values <- p.adjust(p_values, method = "fdr")
Showing the results, I get:
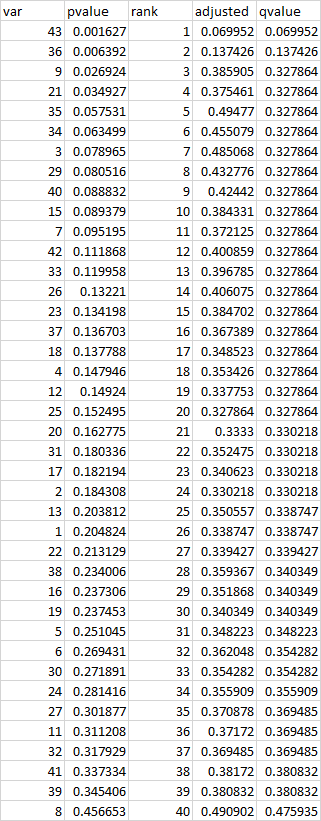
Looking at the table, I would say that after the FDR correction I cannot reject the null hypothesis of no differences between the two groups, for each of the variables. Is it this correct? Otherwise, would it be also true that minimum false discovery rate at which that test may be called significant is 0.069 i.e. for variable 43?
EDIT: at this link http://viiia.org/fdrFigs/?scale=1920 it is stated that 'a q-value measures the False Discovery Rate (FDR) you would incur by accepting the given test and every test with a smaller p-value (and maybe even larger p-values, if they improve the FDR).', so that for my data i would say that for the variable 43 the test has a 6.9% chance of being a false positive. But what about for the variables 9 and 21 for example? Is it correct to say that both the tests have a 32.68% chance of being a false positive? I am wondering if this is the only way of interpreting qvalues or there are more straightforward meanings.
q-valuecolumn is the adjusted p-value. What's in youradjustedcolumn is the adjusted p-value before the correction that forces them to be monotonic with p. $\endgroup$adjustedcolumn is something that i calculated by hand. it was just to understand why then i will get repeated q-values $\endgroup$