There are three important points here. First, the procedure you are using to identify bound proteins is a heuristic approach rather than a formal statistical test. Second, since your data are fundamentally based on rates of radioactive decay, more formal statistical tests might be possible. Third, regardless of whether you stick with heuristics or move to statistical tests, the answer depends on your tradeoffs between false-positive and false-negative results.
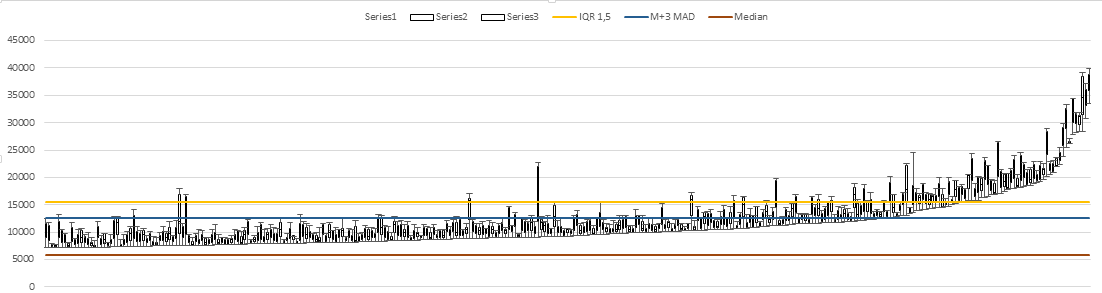
Heuristics. The "median + 3* MAD" approach originates from control-chart analyses formulated nearly a century ago at Bell Labs, in a different context. The original context was to determine when something might be going wrong with a manufacturing process. If values measured on a process were drawn from a normal distribution, then fewer than 3 out of 1000 values are expected to be more than 3 standard deviations (SD) away from the mean value. Even if the underlying distribution is not normal, this "3-sigma" criterion provides a useful guide for prompting an evaluation of whether the "outlier" values represents a process that is out of control. But it's a heuristic guide to further action, not a formal statistical test.
Your application of this heuristic approach is in a different context, as you expect deviations from the center of the distribution to be frequent, with many spots on your array bound by proteins in your lysate. For you, these "outliers" are desirable and are the point of your study. Using the median instead of the mean as the measure of the center of the distribution of values, and the median absolute deviation (MAD*) or the inter-quartile range instead of the SD as a measure of width, avoids some of the complications that thus arise, as these robust measures will be much less affected by the many high values that you expect. For example, in the context of screening all genes to see which ones have significant effects upon knockdown, median-MAD-based heuristics work better than mean-SD for identifying gene candidates for more detailed examination.
Formal statistical tests. In your application you have a pretty good idea of an important underlying statistical process, so you might be able to replace these heuristic assessments with formal statistical tests. Each of your values represents a result of the decay of radioactive phosphorus, a classic example of a Poisson process. A quick look at your data suggests that your values are not decay counts per se, as the within-protein variances seem to exceed the associated mean values as you would expect in a Poisson distribution (the variances do, however, tend to rise with mean values). There is presumably some post-decay detection/amplification process that leads to higher than Poisson variance, in addition to other sources of error such as random differences among the 3 spots for each protein.
A generalized linear model that treats your data as coming from an overdispersed Poisson distribution might lead to an analysis in which standard statistical significance tests are possible, providing within-protein errors (in a log scale for Poisson models) that are normally distributed and independent of the mean values. Modern statistical software provides such analyses readily. This approach has the advantage of removing reliance of the significance of the test result for each individual protein from the vagaries of its own particular 3 values, instead basing the estimate of the underlying measurement error on information pooled from measurements on all 3000+ spots.
False-positives versus false-negatives. Your initial question of whether to stick with the lowest of 3 values for each protein or to use the average in your heuristic assessment of protein binding points out a fundamental decision that you must make. What are your relative risks of making "ON-calls" that are erroneous, versus missing proteins that really are bound but don't pass your criterion for an "ON-call"?
The requirement that the lowest of the 3 values pass something similar to a 3-sigma test is quite stringent, and your top plot suggests that sticking with that requirement will miss a fair number of likely true binding events. Using the average instead, or changing the criterion to something like "median + 2*MAD" will provide more "ON-calls" but runs the risk of making "ON-calls" for more proteins that are not really bound. So there is no single correct answer to that part of your question; you have to decide on your tradeoffs.
If you move to formal statistical tests instead, you do not avoid making this type of decision but you may be able to present your results to others in a more defensible way. You then are faced with the classic multiple comparisons problem. If you perform 1000 significance tests with a criterion of p < 0.05 for significance, your will get about 50 positive results even if there are no true positive cases. There are two general ways of addressing this problem.
You can try to control the familywise error rate, trying to make sure than you will make a false-positive error in only 5% of similar experiments. A highly conservative example is the Bonferroni correction, which in this case would require, for 1000 tests, that you only accept significance of an individual test at p < (0.05/1000) = 0.00005. Less stringent corrections are possible, as described in the linked pages.
Alternatively, you can accept that you are going to make some false-positive calls and try to limit the fraction of such calls among all the calls that you make. This is called controlling the false discovery rate. This is a well-accepted approach for evaluating results of microarrays, of which your application is an example. You might, for example, choose to aim for a 10% false discovery rate, so that about 90% of your "ON-calls" would be true. That could be readily justified in a scientific publication.
* Note that there is an ambiguity in the use of "MAD" even in the context of "median absolute deviation." What is reported as the "MAD" is typically not the median of the absolute values of all deviations from the median, but that result scaled by 1.4826 so that the MAD and the SD should coincide for a normal distribution.