I'm working on a machine learning project involving statistical analysis (and later discriminatory classification) of different proteins (samples) drawn from multiple, potentially overlapping classes / groups, all of which are drawn from a much larger background population (all mammal proteins).
I have a list of features that I calculate for each individual protein, and then serve as the basis for classification (using machine learning) for each class / group of proteins later. (The features are continuous and numerical, but may be very different, and there's no reason to assume that the underlying distribution is normal, or related).
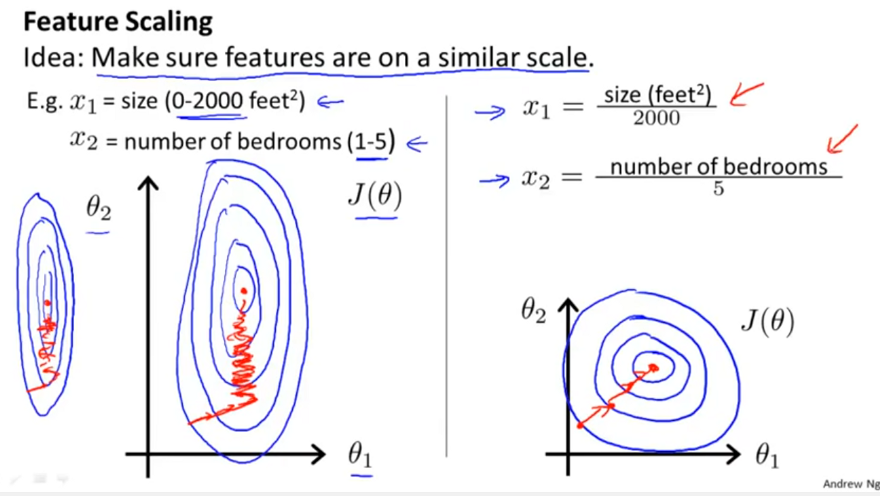
I want to normalize and center the "raw" calculated feature values for later training. The standard approach of normalizing by a Z-score, then centering [0,1] seems inappropiate, since there's no reason to assume the underlying distributions are normal (I have hundreds of different features - frequency counts, bigrams counts, physiochemical property values etc') .
I've heard of "robust statistical measures", and thought of first normalizing (using the medians) all the features against each other, then applying scikit's normalization+centering to the "median normalized" set of features, but I have no idea if this makes sense, or will retain the differences in the original data.
(Note - I also expect a small amount of significant outliers for different features and properties, so using the median is attractive in that regard as well) . Does this make sense? Is there a better way to normalize between all the groups (Rather than just using raw scores for the features)?