To asses differences between two DNA sequencing methods (454 and MiSeq) I have made artificial mixtures of different bacteria in the lab (mock communities).
An Example might be (numbers are percentages of bacterial cells in the sequenced samples):
- Bacterial strain A: 25%
- Bacterial strain B: 25%
- Bacterial strain C: 25%
- Bacterial strain D: 25%
I have also more complex mixtures (up to 20 different bacteria and other ratios than 1:1, for exp. logarithmic)
If you sequence this you get for example the following results:
454:
- A: 20%
- B: 30%
- C: 24%
- D: 26%
Illumina:
- A: 22%
- B: 28%
- C: 20%
- D: 30%
From every sample (like the one above) I have triplicates.
What statistical test would you suggest to test if there is a significant difference between the two methods (I guess analysing the difference in ratios is the right thing?). Is Chi-Square the right thing if I have three different samples (One is the one mentioned above)? Would even some bootstrapping approach be an idea?
I am familiar with R, so if there is any package you can suggest, that would be nice as well...
An image to explain the workflow. I hove this helps to understand my problem better:
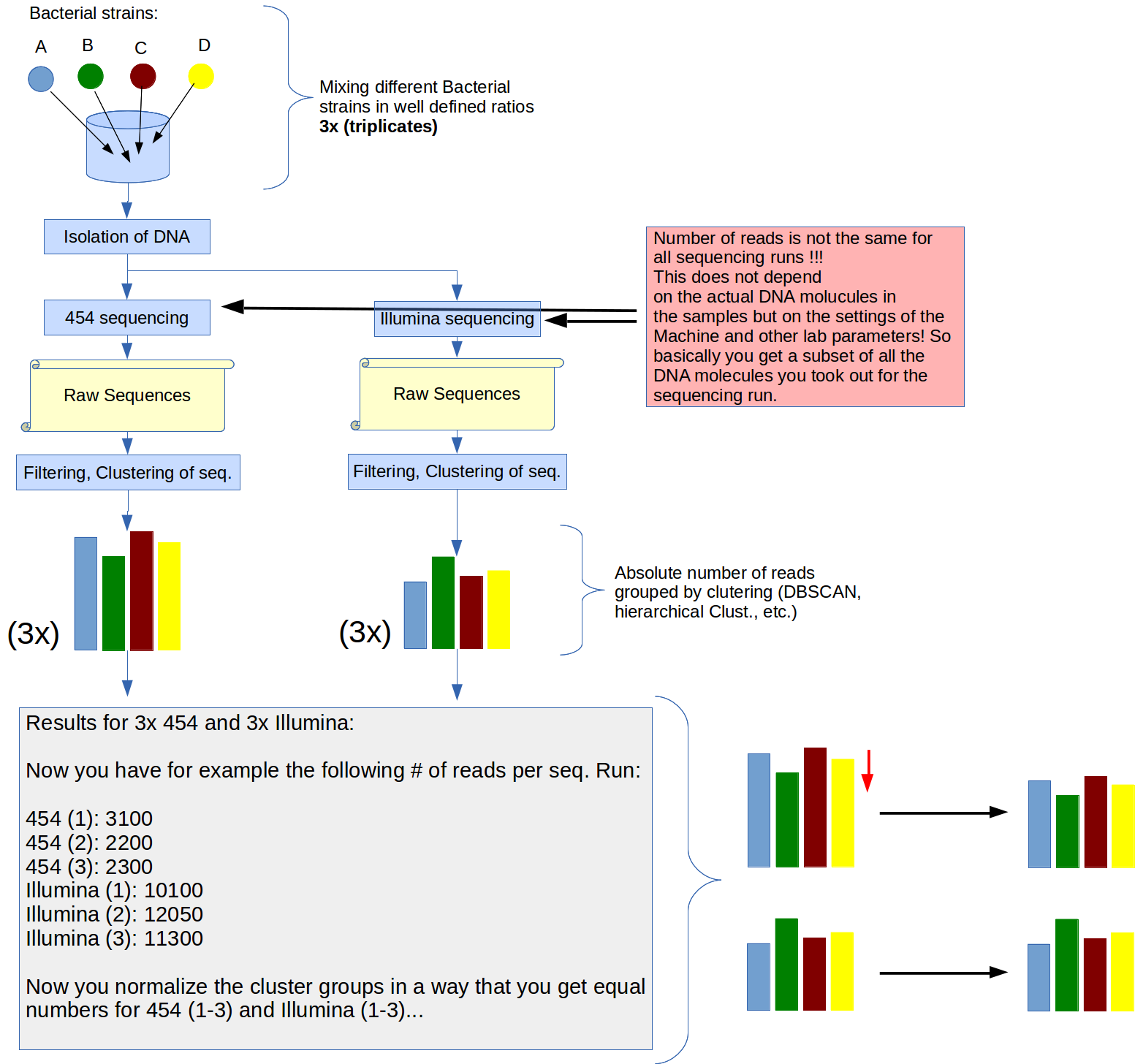
I also have mock communities that are more complex (10 or 19 bacterial strains in different ratios). But the principle is the same.
For the sequencing process you decide how many DNA sequences you want to sequence! The number of reads (Sequences you get) are not (only) a matter of DNA concentration (as long as you have enough DNA to have a "satturation"). So what you can compare at the end between 454 and MiSeq are rather ratios (after clustering) than complete numbers, as far as I understand...