I'm trying some metrics to filter a list of gene names with associated numerical values which correspond to their abundance.
I'm looking for some interesting genes and trying to see if they come up to the top region of the ranked gene list (ranked by abundance)
before and after filtering. Please see Fig. enclosed.
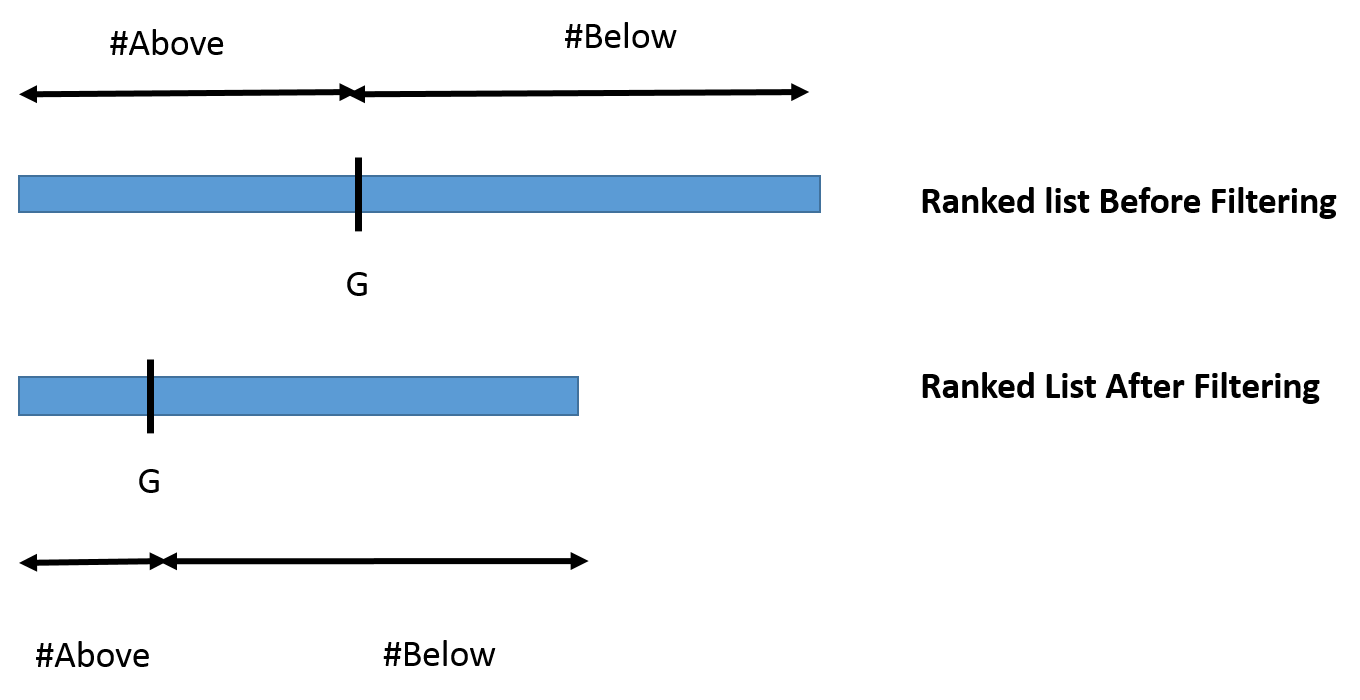
I wish to see if the ranking of my gene of interest (G) has changed significantly before and after filtering and I'm thinking to do a Fisher's Exact test for proportions to ascertain the significance.
So given the rank of the gene of interest I'm willing to look at the number of genes above and below the gene in rank with and without the filtering and compare their proportions using FE Test
If in the UN-Filtered data there are 200 and 2800 genes above and below G and 30 and 100 in the Filtered data respectively then I can do the FE test as follows (in R):
>my.mat <- matrix(c(30,100,200,2800),nrow=2,byrow=TRUE)
>my.mat
[,1] [,2]
[1,] 30 100
[2,] 200 2800
>fisher.test(my.mat,alternative="two.sided")
Please let me know whether I can do a Fisher Exact test AT ALL for such kind of measurement ?
Thanks in advance